How to: assign ChIP-seq peaks to genes
This tutorial demonstrates one way to assign CTCF ChIP-seq peaks to the nearest genes using bioframe.
import matplotlib.pyplot as plt
import numpy as np
import bioframe
base_dir = "/tmp/bioframe_tutorial_data/"
assembly = "hg38"
Load chromosome sizes
chromsizes = bioframe.fetch_chromsizes(assembly)
chromsizes.tail()
name
chr21 46709983
chr22 50818468
chrX 156040895
chrY 57227415
chrM 16569
Name: length, dtype: int64
chromosomes = bioframe.make_viewframe(chromsizes)
Load CTCF ChIP-seq peaks for HFF from ENCODE
This approach makes use of the narrowPeak schema for bioframe.read_table .
ctcf_peaks = bioframe.read_table(
"https://www.encodeproject.org/files/ENCFF401MQL/@@download/ENCFF401MQL.bed.gz",
schema="narrowPeak",
)
ctcf_peaks.head()
| chrom | start | end | name | score | strand | fc | -log10p | -log10q | relSummit | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | chr19 | 48309541 | 48309911 | . | 1000 | . | 5.04924 | -1.0 | 0.00438 | 185 |
| 1 | chr4 | 130563716 | 130564086 | . | 993 | . | 5.05052 | -1.0 | 0.00432 | 185 |
| 2 | chr1 | 200622507 | 200622877 | . | 591 | . | 5.05489 | -1.0 | 0.00400 | 185 |
| 3 | chr5 | 112848447 | 112848817 | . | 869 | . | 5.05841 | -1.0 | 0.00441 | 185 |
| 4 | chr1 | 145960616 | 145960986 | . | 575 | . | 5.05955 | -1.0 | 0.00439 | 185 |
# Filter for selected chromosomes:
ctcf_peaks = bioframe.overlap(ctcf_peaks, chromosomes).dropna(subset=["name_"])[
ctcf_peaks.columns
]
Get list of genes from UCSC
UCSC genes are stored in .gtf format.
genes_url = (
"https://hgdownload.soe.ucsc.edu/goldenpath/hg38/bigZips/genes/hg38.ensGene.gtf.gz"
)
genes = bioframe.read_table(genes_url, schema="gtf").query('feature=="CDS"')
genes.head()
## Note this functions to parse the attributes of the genes:
# import bioframe.sandbox.gtf_io
# genes_attr = bioframe.sandbox.gtf_io.parse_gtf_attributes(genes['attributes'])
| chrom | source | feature | start | end | score | strand | frame | attributes | |
|---|---|---|---|---|---|---|---|---|---|
| 47 | chr1 | ensGene | CDS | 69091 | 70005 | . | + | 0 | gene_id "ENSG00000186092"; transcript_id "ENST... |
| 112 | chr1 | ensGene | CDS | 182709 | 182746 | . | + | 0 | gene_id "ENSG00000279928"; transcript_id "ENST... |
| 114 | chr1 | ensGene | CDS | 183114 | 183240 | . | + | 1 | gene_id "ENSG00000279928"; transcript_id "ENST... |
| 116 | chr1 | ensGene | CDS | 183922 | 184155 | . | + | 0 | gene_id "ENSG00000279928"; transcript_id "ENST... |
| 122 | chr1 | ensGene | CDS | 185220 | 185350 | . | - | 2 | gene_id "ENSG00000279457"; transcript_id "ENST... |
# Filter for selected chromosomes:
genes = bioframe.overlap(genes, chromosomes).dropna(subset=["name_"])[genes.columns]
Assign each peak to the gene


Here, we want to assign each peak (feature) to a gene (input table).
peaks_closest = bioframe.closest(genes, ctcf_peaks)
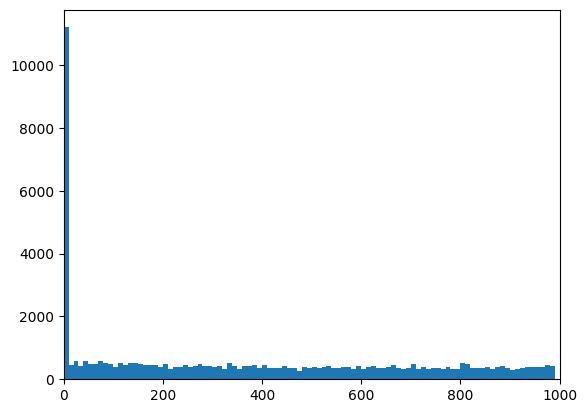
# Plot the distribution of distances from peaks to genes:
plt.hist(peaks_closest["distance"], np.arange(0, 1e3, 10))
plt.xlim([0, 1e3])
(0.0, 1000.0)
Ignore upstream/downstream peaks from genes (strand-indifferent version)
Sometimes you may want to ignore all the CTCFs upstream from the genes.
By default, bioframe.overlap does not know the orintation of the genes, and thus assumes that the upstream/downstream is defined by the genomic coordinate (upstream is the direction towards the smaller coordinate):
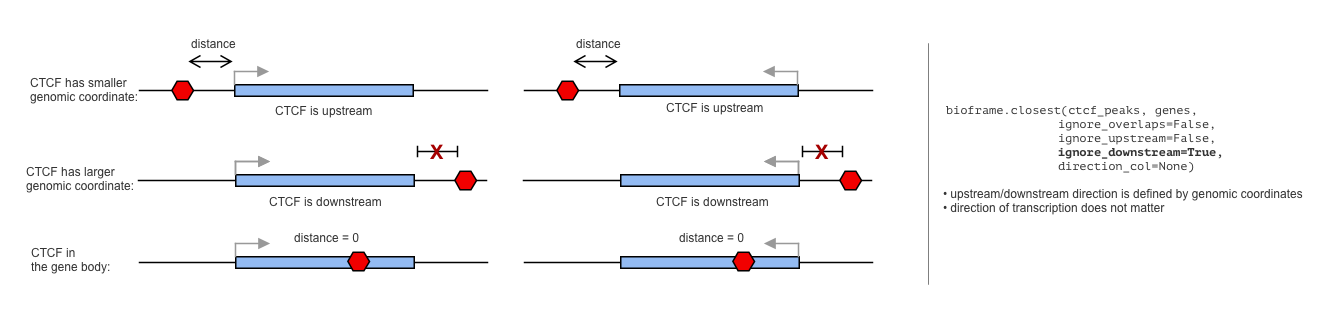
peaks_closest_upstream_nodir = bioframe.closest(
genes,
ctcf_peaks,
ignore_overlaps=False,
ignore_upstream=False,
ignore_downstream=True,
direction_col=None,
)
peaks_closest_downstream_nodir = bioframe.closest(
genes,
ctcf_peaks,
ignore_overlaps=False,
ignore_upstream=True,
ignore_downstream=False,
direction_col=None,
)
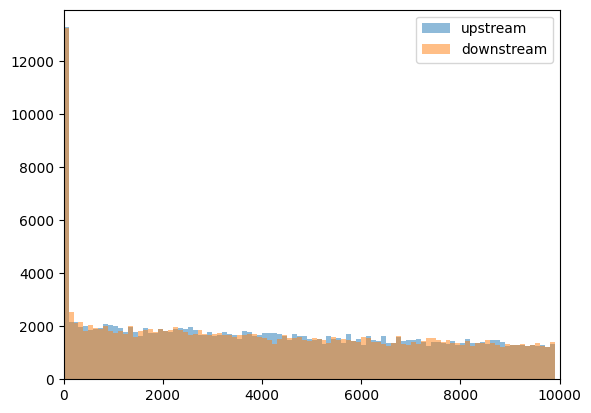
Note that distribution did not change much, and upstream and downstream distances are very similar:
plt.hist(
peaks_closest_upstream_nodir["distance"],
np.arange(0, 1e4, 100),
alpha=0.5,
label="upstream",
)
plt.hist(
peaks_closest_downstream_nodir["distance"],
np.arange(0, 1e4, 100),
alpha=0.5,
label="downstream",
)
plt.xlim([0, 1e4])
plt.legend()
<matplotlib.legend.Legend at 0x7e82d6a4c1a0>
Ignore upstream/downstream peaks from genes (strand-aware version)
More biologically relevant approach will be to define upstream/downstream by strand of the gene. CTCF upstream of transcription start site might play different role than CTCF after transcription end site.
bioframe.closest has the parameter direction_col to control for that:
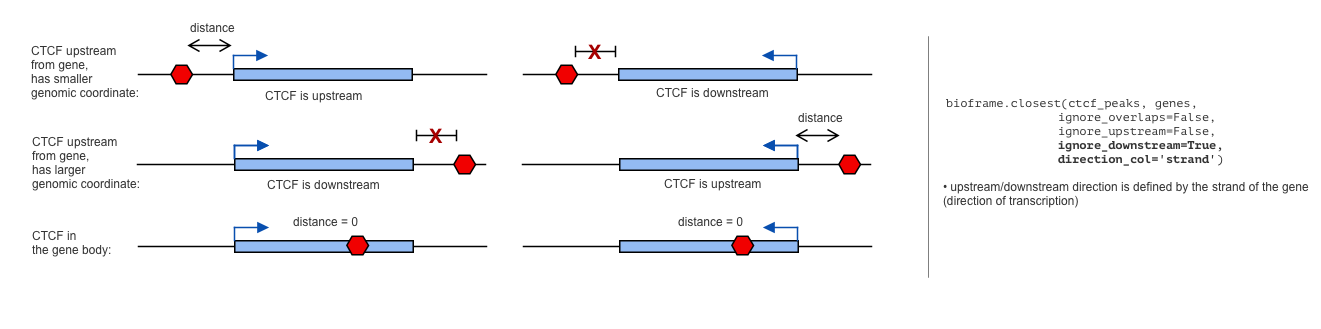
# Note that "strand" here is the column name in genes table:
peaks_closest_upstream_dir = bioframe.closest(
genes,
ctcf_peaks,
ignore_overlaps=False,
ignore_upstream=False,
ignore_downstream=True,
direction_col="strand",
)
peaks_closest_downstream_dir = bioframe.closest(
genes,
ctcf_peaks,
ignore_overlaps=False,
ignore_upstream=True,
ignore_downstream=False,
direction_col="strand",
)
plt.hist(
peaks_closest_upstream_dir["distance"],
np.arange(0, 1e4, 100),
alpha=0.5,
label="upstream",
)
plt.hist(
peaks_closest_downstream_dir["distance"],
np.arange(0, 1e4, 100),
alpha=0.5,
label="downstream",
)
plt.xlim([0, 1e4])
plt.legend()
<matplotlib.legend.Legend at 0x7e82ce690410>
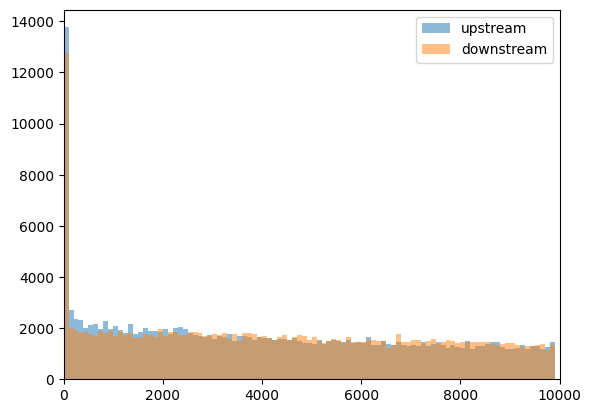
CTCF peaks upstream of the genes are more enriched at short distances to TSS, if we take the strand into account.