Performance
This notebook illustrates performance of typical use cases for bioframe on sets of randomly generated intervals.
# ! pip install pyranges
## Optional:
# ! conda install -c bioconda bedtools
# ! pip install pybedtools
import platform
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import psutil
import pyranges
import bioframe
plt.rcParams["figure.facecolor"] = "white"
plt.rcParams["font.size"] = 16
# Note that by default we switch off the demo of pybedtools.
# It runs for minutes for 10^5 intervals
include_pybedtools = False
if include_pybedtools:
import pybedtools
pybedtools.helpers.set_bedtools_path(
path="/usr/bin/"
) # Set the path to bedtools CLI
print(f"Bioframe v.{bioframe.__version__}")
print(f"PyRanges v.{pyranges.__version__}")
if include_pybedtools:
print(f"Pybedtools v.{pybedtools.__version__}")
print(f"System Platform: {platform.platform()}")
print(f"{psutil.cpu_count()} CPUs at {psutil.cpu_freq().current:.0f} GHz")
Bioframe v.0.5.1
PyRanges v.0.0.129
System Platform: Linux-5.19.0-46-generic-x86_64-with-glibc2.35
24 CPUs at 3040 GHz
Below we define a function to generate random intervals with various properties, returning a dataframe of intervals.
def make_random_intervals(
n=1e5,
n_chroms=1,
max_coord=None,
max_length=10,
sort=False,
categorical_chroms=False,
):
n = int(n)
n_chroms = int(n_chroms)
max_coord = (n // n_chroms) if max_coord is None else int(max_coord)
max_length = int(max_length)
chroms = np.array(["chr" + str(i + 1) for i in range(n_chroms)])[
np.random.randint(0, n_chroms, n)
]
starts = np.random.randint(0, max_coord, n)
ends = starts + np.random.randint(1, max_length, n)
df = pd.DataFrame({"chrom": chroms, "start": starts, "end": ends})
if categorical_chroms:
df["chrom"] = df["chrom"].astype("category")
if sort:
df = df.sort_values(["chrom", "start", "end"]).reset_index(drop=True)
return df
Overlap
In this chapter we characterize the performance of the key function, bioframe.overlap. We show that the speed depends on:
the number of intervals
number of intersections (or density of intervals)
type of overlap (inner, outer, left)
dtype of chromosomes
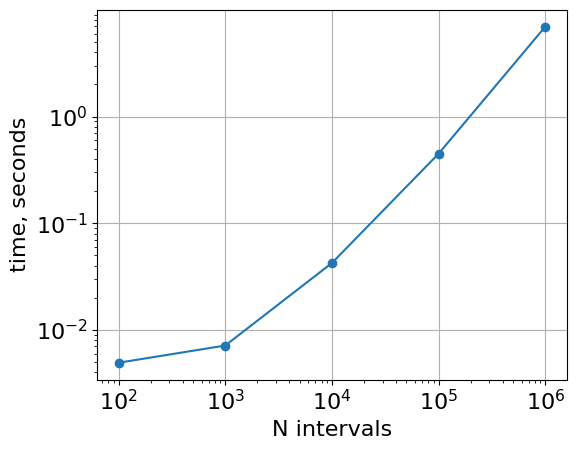
vs number of intervals
timings = {}
for n in [1e2, 1e3, 1e4, 1e5, 1e6]:
df = make_random_intervals(n=n, n_chroms=1)
df2 = make_random_intervals(n=n, n_chroms=1)
timings[n] = %timeit -o -r 1 bioframe.overlap(df, df2)
4.92 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 100 loops each)
7.13 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 100 loops each)
42.3 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
448 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
6.95 s ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
plt.loglog(
list(timings.keys()),
list([r.average for r in timings.values()]),
"o-",
)
plt.xlabel("N intervals")
plt.ylabel("time, seconds")
plt.gca().set_aspect(1.0)
plt.grid()
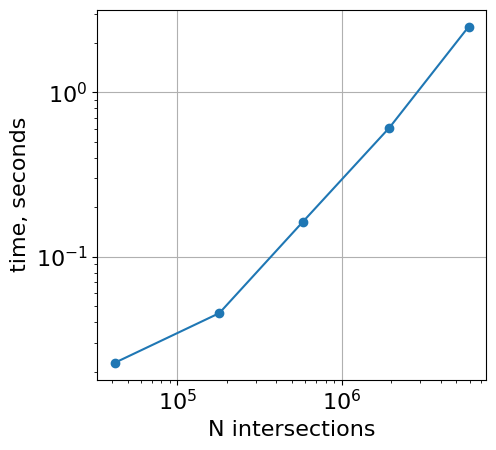
vs total number of intersections
Note that not only the number of intervals, but also the density of intervals determines the performance of overlap.
timings = {}
n_intersections = {}
n = 1e4
for avg_interval_len in [3, 1e1, 3e1, 1e2, 3e2]:
df = make_random_intervals(n=n, n_chroms=1, max_length=avg_interval_len * 2)
df2 = make_random_intervals(n=n, n_chroms=1, max_length=avg_interval_len * 2)
timings[avg_interval_len] = %timeit -o -r 1 bioframe.overlap(df, df2)
n_intersections[avg_interval_len] = bioframe.overlap(df, df2).shape[0]
22.7 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
45.5 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
163 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
611 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
2.51 s ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
plt.loglog(
list(n_intersections.values()),
list([r.average for r in timings.values()]),
"o-",
)
plt.xlabel("N intersections")
plt.ylabel("time, seconds")
plt.gca().set_aspect(1.0)
plt.grid()
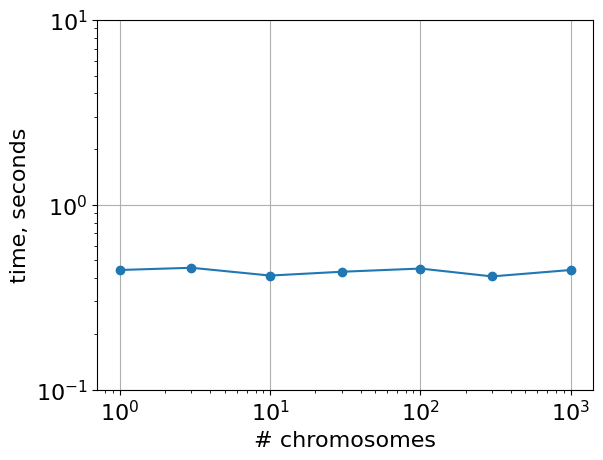
vs number of chromosomes
If we consider a genome of the same length, divided into more chromosomes, the timing is relatively unaffected.
timings = {}
n_intersections = {}
n = 1e5
for n_chroms in [1, 3, 10, 30, 100, 300, 1000]:
df = make_random_intervals(n, n_chroms)
df2 = make_random_intervals(n, n_chroms)
timings[n_chroms] = %timeit -o -r 1 bioframe.overlap(df, df2)
n_intersections[n_chroms] = bioframe.overlap(df, df2).shape[0]
443 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
456 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
414 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
434 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
451 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
409 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
443 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
Note this test preserves the number of intersections, which is likely why performance remains similar over the considered range.
n_intersections
{1: 810572,
3: 810871,
10: 809463,
30: 815322,
100: 808166,
300: 802130,
1000: 787235}
plt.loglog(
list(timings.keys()),
list([r.average for r in timings.values()]),
"o-",
)
plt.ylim([1e-1, 10])
plt.xlabel("# chromosomes")
plt.ylabel("time, seconds")
# plt.gca().set_aspect(1.0)
plt.grid()
vs other parameters: join type, sorted or categorical inputs
Note that default for overlap: how='left', keep_order=True, and the returned dataframe is sorted after the overlaps have been ascertained. Also note that keep_order=True is only a valid argument for how='left' as the order is not well-defined for inner or outer overlaps.
df = make_random_intervals()
df2 = make_random_intervals()
%timeit -r 1 bioframe.overlap(df, df2)
%timeit -r 1 bioframe.overlap(df, df2, how='left', keep_order=False)
418 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
274 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
df = make_random_intervals()
df2 = make_random_intervals()
%timeit -r 1 bioframe.overlap(df, df2, how='outer')
%timeit -r 1 bioframe.overlap(df, df2, how='inner')
%timeit -r 1 bioframe.overlap(df, df2, how='left', keep_order=False)
329 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
151 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
247 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
Note below that detection of overlaps takes a relatively small fraction of the execution time. The majority of the time the user-facing function spends on formatting the output table.
df = make_random_intervals()
df2 = make_random_intervals()
%timeit -r 1 bioframe.overlap(df, df2)
%timeit -r 1 bioframe.overlap(df, df2, how='inner')
%timeit -r 1 bioframe.ops._overlap_intidxs(df, df2)
%timeit -r 1 bioframe.ops._overlap_intidxs(df, df2, how='inner')
449 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
148 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
61 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
62.4 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
Note that sorting inputs provides a moderate speedup, as well as storing chromosomes as categoricals
print("Default inputs (outer/inner joins):")
df = make_random_intervals()
df2 = make_random_intervals()
%timeit -r 1 bioframe.overlap(df, df2)
%timeit -r 1 bioframe.overlap(df, df2, how='inner')
print("Sorted inputs (outer/inner joins):")
df_sorted = make_random_intervals(sort=True)
df2_sorted = make_random_intervals(sort=True)
%timeit -r 1 bioframe.overlap(df_sorted, df2_sorted)
%timeit -r 1 bioframe.overlap(df_sorted, df2_sorted, how='inner')
print("Categorical chromosomes (outer/inner joins):")
df_cat = make_random_intervals(categorical_chroms=True)
df2_cat = make_random_intervals(categorical_chroms=True)
%timeit -r 1 bioframe.overlap(df_cat, df2_cat)
%timeit -r 1 bioframe.overlap(df_cat, df2_cat, how='inner')
Default inputs (outer/inner joins):
440 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
149 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
Sorted inputs (outer/inner joins):
331 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
137 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
Categorical chromosomes (outer/inner joins):
333 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
90 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
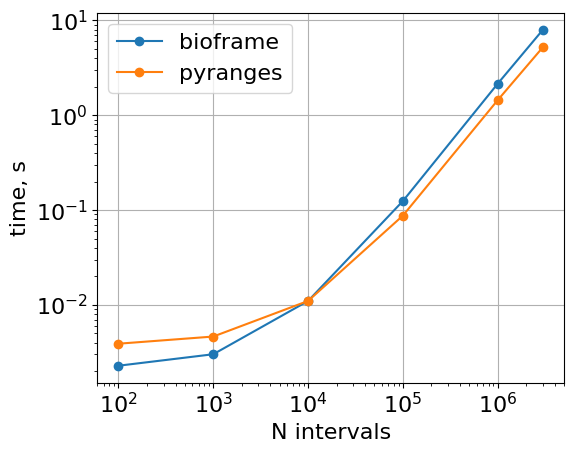
Vs Pyranges (and, optionally, pybedtools)
Default arguments
The core intersection function of PyRanges is faster, since PyRanges object splits intervals by chromosomes at the object construction stage
def df2pr(df):
return pyranges.PyRanges(
chromosomes=df.chrom,
starts=df.start,
ends=df.end,
)
timings_bf = {}
timings_pr = {}
for n in [1e2, 1e3, 1e4, 1e5, 1e6, 3e6]:
df = make_random_intervals(n=n, n_chroms=1)
df2 = make_random_intervals(n=n, n_chroms=1)
pr = df2pr(df)
pr2 = df2pr(df2)
timings_bf[n] = %timeit -o -r 1 bioframe.overlap(df, df2,how='inner')
timings_pr[n] = %timeit -o -r 1 pr.join(pr2)
2.36 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 100 loops each)
1.49 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1,000 loops each)
2.94 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 100 loops each)
1.9 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1,000 loops each)
10.8 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 100 loops each)
6.63 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 100 loops each)
128 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
69.4 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
2.35 s ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
1.16 s ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
7.97 s ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
4.75 s ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
plt.loglog(
list(timings_bf.keys()),
list([r.average for r in timings_bf.values()]),
"o-",
label="bioframe",
)
plt.loglog(
list(timings_pr.keys()),
list([r.average for r in timings_pr.values()]),
"o-",
label="pyranges",
)
plt.gca().set(
xlabel="N intervals",
ylabel="time, seconds",
aspect=1.0,
xticks=10 ** np.arange(2, 6.1),
)
plt.grid()
plt.legend()
<matplotlib.legend.Legend at 0x7fe3556e94b0>
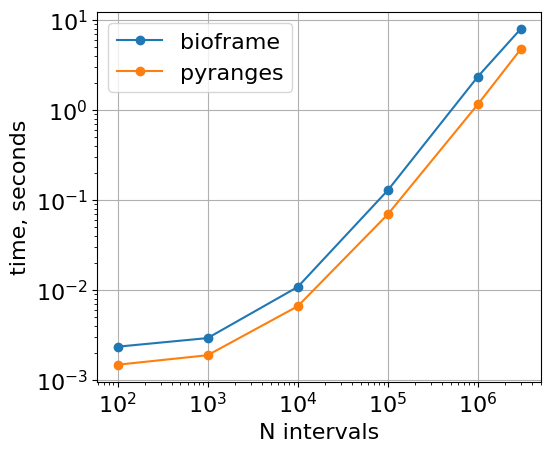
With roundtrips to dataframes
Note that pyranges performs useful calculations at the stage of creating a PyRanges object. Thus a direct comparison for one-off operations on pandas DataFrames between bioframe and pyranges should take this step into account. This roundrip is handled by pyranges_intersect_dfs below.
def pyranges_intersect_dfs(df, df2):
return df2pr(df).intersect(df2pr(df2)).as_df()
if include_pybedtools:
def pybedtools_intersect_dfs(bed1, bed2):
return bed1.intersect(bed2).to_dataframe()
timings_bf = {}
timings_pr = {}
if include_pybedtools:
timings_pb = {}
for n in [1e2, 1e3, 1e4, 1e5, 1e6, 3e6]:
df = make_random_intervals(n=n, n_chroms=1)
df2 = make_random_intervals(n=n, n_chroms=1)
timings_bf[n] = %timeit -o -r 1 bioframe.overlap(df, df2, how='inner')
timings_pr[n] = %timeit -o -r 1 pyranges_intersect_dfs(df, df2)
if include_pybedtools:
bed1 = pybedtools.BedTool.from_dataframe(df)
bed2 = pybedtools.BedTool.from_dataframe(df2)
timings_pb[n] = %timeit -o -r 1 pybedtools_intersect_dfs(bed1, bed2)
2.28 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 100 loops each)
3.9 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 100 loops each)
3.02 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 100 loops each)
4.64 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 100 loops each)
11 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 100 loops each)
11 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 100 loops each)
125 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
87.4 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
2.15 s ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
1.44 s ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
7.98 s ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
5.23 s ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
plt.loglog(
list(timings_bf.keys()),
list([r.average for r in timings_bf.values()]),
"o-",
label="bioframe",
)
plt.loglog(
list(timings_pr.keys()),
list([r.average for r in timings_pr.values()]),
"o-",
label="pyranges",
)
if include_pybedtools:
plt.loglog(
list(timings_pb.keys()),
list([r.average for r in timings_pb.values()]),
"o-",
label="pybedtools",
)
plt.gca().set(xlabel="N intervals", ylabel="time, seconds", aspect=1.0)
plt.grid()
plt.legend()
<matplotlib.legend.Legend at 0x7fe355f4d420>
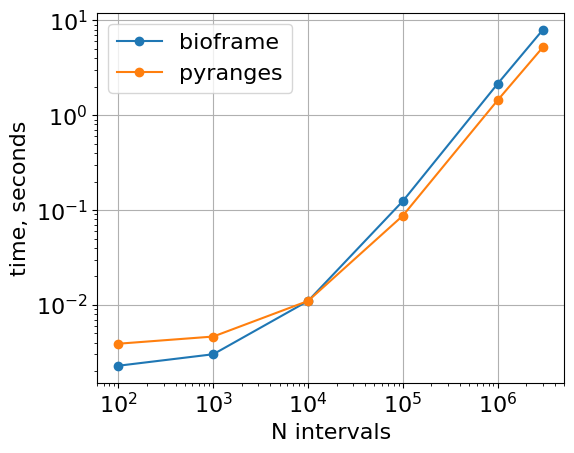
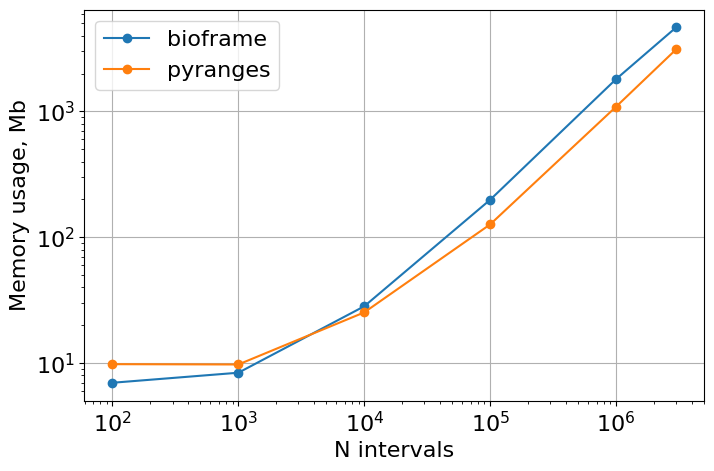
Memory usage
import time
from memory_profiler import memory_usage
def sleep_before_after(func, sleep_sec=0.5):
"""
Wrapper that allows to report background interpreter's memory consumption
for the first 5 time intervals (if increment is 0.1 abd sleep_sec=0.5):
https://github.com/pythonprofilers/memory_profiler#api
"""
def _f(*args, **kwargs):
time.sleep(sleep_sec)
func(*args, **kwargs)
time.sleep(sleep_sec)
return _f
mem_usage_bf = {}
mem_usage_pr = {}
if include_pybedtools:
mem_usage_pb = {}
for n in [1e2, 1e3, 1e4, 1e5, 1e6, 3e6]:
df = make_random_intervals(n=n, n_chroms=1)
df2 = make_random_intervals(n=n, n_chroms=1)
mem_usage_bf[n] = memory_usage(
(sleep_before_after(bioframe.overlap), (df, df2), dict(how="inner")),
backend="psutil_pss",
include_children=True,
interval=0.1,
)
mem_usage_pr[n] = memory_usage(
(sleep_before_after(pyranges_intersect_dfs), (df, df2), dict()),
backend="psutil_pss",
include_children=True,
interval=0.1,
)
if include_pybedtools:
bed1 = pybedtools.BedTool.from_dataframe(df)
bed2 = pybedtools.BedTool.from_dataframe(df2)
mem_usage_pb[n] = memory_usage(
(sleep_before_after(pybedtools_intersect_dfs), (bed1, bed2), dict()),
backend="psutil_pss",
include_children=True,
interval=0.1,
)
# Note that r[4] is the background memory usage of Python interpreter,
# and max(r) is the maximum memory usage (that must be from the
# bioframe/pyranges functions)
plt.figure(figsize=(8, 6))
plt.loglog(
list(mem_usage_bf.keys()),
list([max(r) - r[4] for r in mem_usage_bf.values()]),
"o-",
label="bioframe",
)
plt.loglog(
list(mem_usage_pr.keys()),
list([max(r) - r[4] for r in mem_usage_pr.values()]),
"o-",
label="pyranges",
)
if include_pybedtools:
plt.loglog(
list(mem_usage_pb.keys()),
list([max(r) - r[4] for r in mem_usage_pb.values()]),
"o-",
label="pybedtools",
)
plt.gca().set(xlabel="N intervals", ylabel="Memory usage, Mb", aspect=1.0)
plt.grid()
plt.legend()
<matplotlib.legend.Legend at 0x7fe38f4014b0>
print("Bioframe dtypes:")
display(df.dtypes)
print()
print("Pyranges dtypes:")
display(df2pr(df).dtypes)
if include_pybedtools:
print("Pybedtools dtypes:")
bed1 = pybedtools.BedTool.from_dataframe(df)
display(bed1.to_dataframe().dtypes)
Bioframe dtypes:
chrom object
start int64
end int64
dtype: object
Pyranges dtypes:
Chromosome category
Start int64
End int64
dtype: object
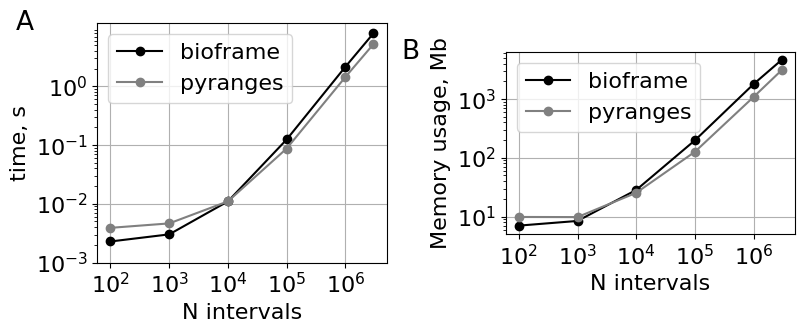
### Combined performance figure.
fig, axs = plt.subplot_mosaic("AAA.BBB", figsize=(9.0, 4))
plt.sca(axs["A"])
plt.text(
-0.25,
1.0,
"A",
horizontalalignment="center",
verticalalignment="center",
transform=plt.gca().transAxes,
fontsize=19,
)
plt.loglog(
list(timings_bf.keys()),
list([r.average for r in timings_bf.values()]),
"o-",
color="k",
label="bioframe",
)
plt.loglog(
list(timings_pr.keys()),
list([r.average for r in timings_pr.values()]),
"o-",
color="gray",
label="pyranges",
)
if include_pybedtools:
plt.loglog(
list(timings_pb.keys()),
list([r.average for r in timings_pb.values()]),
"o-",
color="lightgray",
label="pybedtools",
)
plt.gca().set(
xlabel="N intervals",
ylabel="time, s",
aspect=1.0,
xticks=10 ** np.arange(2, 6.1),
yticks=10 ** np.arange(-3, 0.1),
)
plt.grid()
plt.legend()
plt.sca(axs["B"])
plt.text(
-0.33,
1.0,
"B",
horizontalalignment="center",
verticalalignment="center",
transform=plt.gca().transAxes,
fontsize=19,
)
plt.loglog(
list(mem_usage_bf.keys()),
list([max(r) - r[4] for r in mem_usage_bf.values()]),
"o-",
color="k",
label="bioframe",
)
plt.loglog(
list(mem_usage_pr.keys()),
list([max(r) - r[4] for r in mem_usage_pr.values()]),
"o-",
color="gray",
label="pyranges",
)
if include_pybedtools:
plt.loglog(
list(mem_usage_pb.keys()),
list([max(r) - r[4] for r in mem_usage_pb.values()]),
"o-",
color="lightgray",
label="pybedtools",
)
plt.gca().set(
xlabel="N intervals",
ylabel="Memory usage, Mb",
aspect=1.0,
xticks=10 ** np.arange(2, 6.1),
)
plt.grid()
plt.legend()
<matplotlib.legend.Legend at 0x7fe3548d5120>
Slicing
timings_slicing_bf = {}
timings_slicing_pr = {}
for n in [1e2, 1e3, 1e4, 1e5, 1e6, 3e6]:
df = make_random_intervals(n=n, n_chroms=1)
timings_slicing_bf[n] = %timeit -o -r 1 bioframe.select(df, ('chr1', n//2, n//4*3))
pr = df2pr(df)
timings_slicing_pr[n] = %timeit -o -r 1 pr['chr1', n//2:n//4*3]
334 µs ± 0 ns per loop (mean ± std. dev. of 1 run, 1,000 loops each)
468 µs ± 0 ns per loop (mean ± std. dev. of 1 run, 1,000 loops each)
346 µs ± 0 ns per loop (mean ± std. dev. of 1 run, 1,000 loops each)
593 µs ± 0 ns per loop (mean ± std. dev. of 1 run, 1,000 loops each)
668 µs ± 0 ns per loop (mean ± std. dev. of 1 run, 1,000 loops each)
1.92 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1,000 loops each)
3.54 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 100 loops each)
18.1 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 100 loops each)
40.1 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
222 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
118 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 10 loops each)
804 ms ± 0 ns per loop (mean ± std. dev. of 1 run, 1 loop each)
plt.loglog(
list(timings_slicing_bf.keys()),
list([r.average for r in timings_bf.values()]),
"o-",
label="bioframe",
)
plt.loglog(
list(timings_slicing_pr.keys()),
list([r.average for r in timings_pr.values()]),
"o-",
label="pyranges",
)
plt.gca().set(xlabel="N intervals", ylabel="time, s", aspect=1.0)
plt.grid()
plt.legend()
<matplotlib.legend.Legend at 0x7fe3174e49d0>